La maladie de Pompe ou la maladie du stockage du glycogène II (GSD-II)
La maladie de Pompe, également connue sous le nom de maladie du stockage de glycogène II (GSD-II) ou déficit en maltase acide, est l’un des 49 troubles lysosomaux connus.
Le nom de la maladie de Pompe vient du pathologiste néerlandais J.C. Pompe, qui a décrit pour la première fois un nourrisson atteint de la maladie en 1932. La maladie de Pompe affecte environ 5 000 à 10 000 personnes dans le monde. Aux États-Unis, on estime qu’il touche 1 personne sur 40 000.
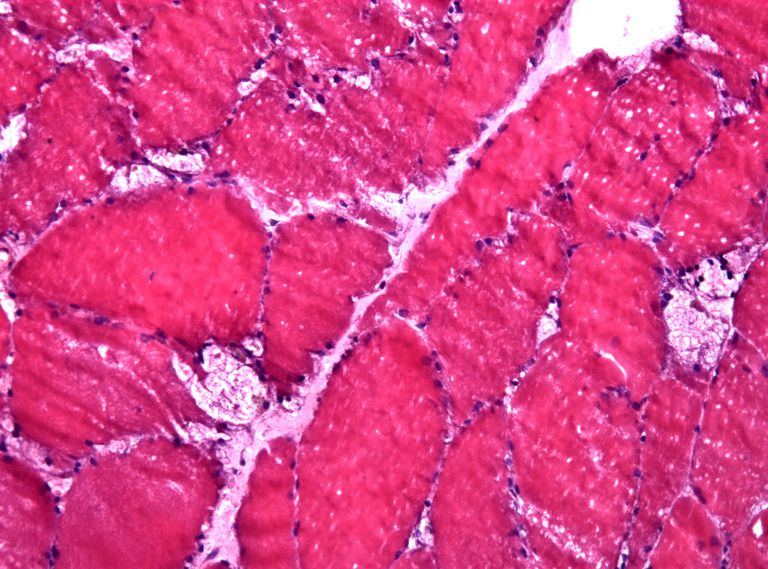
La maladie de Pompe est causée par une carence ou l’absence complète d’une enzyme appelée acide alpha-glucosidase. Si cette enzyme ne fonctionne pas correctement, le glycogène, un sucre complexe, s’accumule dans les cellules du corps et cause des dommages aux organes et aux tissus. Cette accumulation affecte principalement les muscles du corps, entraînant une faiblesse musculaire généralisée. Cette déficience enzymatique peut devenir mortelle lorsque les muscles respiratoires et cardiaques sont affectés. La condition est génétique, et les deux parents doivent porter le gène muté pour que leur enfant en hérite.
Il existe deux formes de la maladie de Pompe – apparition infantile et apparition tardive – qui provoquent toutes deux une faiblesse musculaire. La progression de la maladie dépend du début de la maladie.
Maladie de Pompe Infantile-Survenue
Infantile-apparition est considérée comme la forme grave de la maladie de Pompe. La condition apparaît habituellement dans les premiers mois de la vie. Les nourrissons sont faibles et ont du mal à tenir la tête. Leurs muscles cardiaques deviennent malades et leurs coeurs deviennent élargis et faibles. Ils peuvent également avoir de grandes langues saillantes et un foie élargi.
Autres symptômes:
Insuffisance de croissance et prise de poids (retard de croissance)
- Anomalies cardiaques et rythme cardiaque irrégulier
- Difficulté respiratoire pouvant inclure des évanouissements
- Problèmes d’alimentation et de déglutition mil Absence de jalons de développement comme le retournement ou le ramper
- Problèmes jambes
- Perte auditive
- La maladie progresse rapidement, et les enfants meurent généralement d’insuffisance cardiaque et de faiblesse respiratoire avant leur premier anniversaire. Les enfants affectés peuvent vivre plus longtemps avec des interventions médicales appropriées. Pomp La maladie de Pompe tardive
- La maladie de Pompe à début tardif débute généralement par des symptômes de faiblesse musculaire qui peuvent se manifester à tout moment, de la petite enfance jusqu’à l’âge adulte. La faiblesse musculaire affecte la moitié inférieure du corps plus que les membres supérieurs. La maladie progresse plus lentement que la forme infantile, mais les individus ont encore une espérance de vie réduite.
L’espérance de vie dépend du début de la maladie et de la vitesse à laquelle les symptômes progressent. Les symptômes tels que la difficulté à marcher ou à monter les escaliers commencent et progressent lentement au fil des ans. Comme dans les cas d’apparition précoce, les personnes ayant une apparition tardive peuvent également développer des problèmes respiratoires. À mesure que la maladie progresse, les personnes deviennent dépendantes d’un fauteuil roulant ou alitées et peuvent avoir besoin d’un respirateur pour respirer.
Diagnostic
La maladie de Pompe est généralement diagnostiquée après la progression des symptômes. Chez les adultes, la maladie de Pompe peut être confondue avec d’autres maladies musculaires chroniques telles que la sclérose en plaques. Si votre médecin soupçonne la maladie de Pompe, il peut examiner l’activité de l’enzyme alpha-glucosidase acide dans les cellules cutanées cultivées. Chez les adultes, un test sanguin peut être utilisé pour déterminer une réduction ou une absence de cette enzyme.
Traitement
Une personne atteinte de la maladie de Pompe aura besoin de soins médicaux spécialisés de la part de généticiens, de spécialistes du métabolisme et de neurologues. De nombreuses personnes trouvent que le régime riche en protéines est utile, de même que de nombreux exercices quotidiens.
Des évaluations médicales fréquentes sont nécessaires à mesure que la maladie progresse.
