Avec le dépistage néonatal, cependant, la mucoviscidose est diagnostiquée dans les premières semaines de la vie et les bébés peuvent commencer le traitement immédiatement, évitant ainsi la malnutrition et la détresse respiratoire qui seraient autrement inévitables. Diagnosis Le diagnostic précoce et le traitement ont également le potentiel d’augmenter l’espérance de vie des personnes fibro-kystiques. Les personnes vivant actuellement avec la fibrose kystique peuvent raisonnablement espérer vivre jusqu’à la mi-trentaine, mais certaines études prédisent que les bébés nés aujourd’hui atteints de mucoviscidose vivront dans la cinquantaine en raison d’un diagnostic précoce et d’un meilleur traitement.
Processus de dépistage
Le dépistage néonatal de la fibrose kystique est un test permettant de détecter la «possibilité» d’avoir une fibrose kystique. C’est la première étape d’un processus qui mène finalement à un diagnostic de fibrose kystique, à l’identification des porteurs de CF, ou élimine les deux possibilités. Si les résultats du dépistage initial sont positifs, cela ne signifie pas qu’un bébé a une fibrose kystique. Un dépistage positif signifie simplement que d’autres tests doivent être effectués pour déterminer l’importance du résultat positif.
Première étape – Élever le drapeau rouge
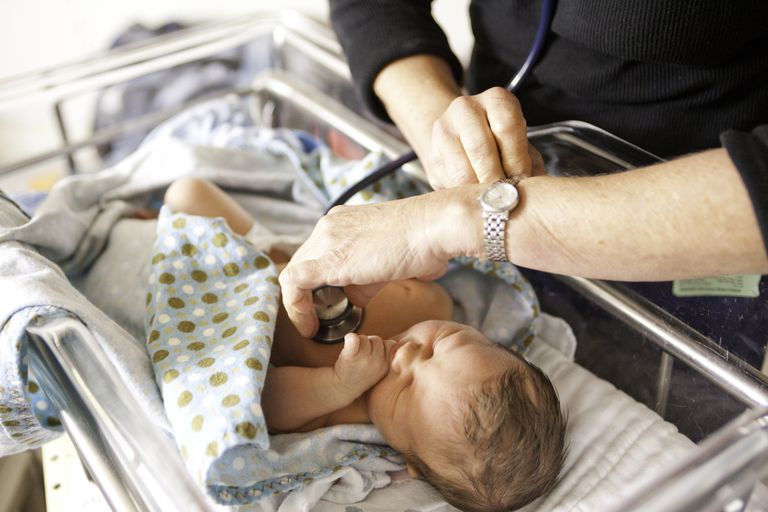
La première étape du dépistage néonatal est un test sanguin effectué quelques jours après la naissance. Du sang est prélevé sur le bébé et envoyé dans un laboratoire d’état pour dépister plusieurs troubles. Le test de dépistage de la mucoviscidose recherche des taux élevés d’une substance appelée trypsinogène immunoréactif (IRT), qui est une enzyme créée par le pancréas. Les bébés nés avec une fibrose kystique ont souvent des niveaux élevés d’IRT dans leur sang, mais d’autres conditions peuvent également entraîner une élévation de l’enzyme. Si un dépistage néonatal de la FK est positif, cela signifie que des taux élevés d’IRT ont été détectés. Chaque état détermine ses propres directives pour les niveaux normaux et anormaux de TRI. Certains états ont une valeur fixe qui constitue un niveau positif et certains états choisissent un pourcentage des niveaux d’IRT les plus élevés rapportés chaque jour.
Deuxième étape – Tests génétiques
Si le dépistage initial du TIR chez les nouveau-nés est positif, la plupart des États effectuent un autre test sanguin pour déterminer si le bébé a une mutation du régulateur de la conductance transmembranaire de la fibrose kystique (CFTR). Il y a plus de 1 200 mutations connues dans le gène CFTR qui causent la fibrose kystique. Il n’est ni pratique, ni financièrement possible de les tester tous, mais la plupart des États testent plusieurs des mutations les plus courantes. Encore une fois, chaque état décide quelles mutations inclure dans le panneau de test.
Si une mutation du gène CFTR est détectée, le bébé est porteur de mucoviscidose ou atteint de la maladie de la fibrose kystique. Le laboratoire de l’État informera le médecin principal du bébé et dans certains États, il avisera également le département de la santé du comté ou un autre organisme approuvé et formé pour assurer le suivi auprès des familles.
Si les taux d’IRT sont élevés mais qu’aucune mutation du gène CFTR n’est détectée, les résultats des deux tests sont envoyés au médecin de soins primaires du bébé qui déterminera si d’autres tests sont nécessaires.
Troisième étape – Test de la sueur
Le test de la sueur au chlorure, ou test de la sueur, a été le test de référence utilisé pour diagnostiquer la fibrose kystique pendant de nombreuses années. Le test mesure la quantité de sel dans la transpiration d’une personne, ce qui est plus élevé que la normale chez les personnes atteintes de mucoviscidose. Une teneur en chlorure supérieure à 60 mmol / litre est considérée comme un résultat positif.
Lorsque le médecin de soins primaires reçoit les résultats du dépistage néonatal du laboratoire d’état, il ou elle décide si d’autres tests doivent être effectués. Si les taux d’IRT étaient élevés mais qu’aucune mutation CFTR n’avait été détectée, le médecin traitant peut de toute façon demander un test de la sueur au cas où le bébé présente l’une des mutations les moins courantes qui ne figurait pas dans le panel de tests génétiques.
Si le dépistage chez le nouveau-né a détecté une mutation CFTR, le médecin de premier recours ordonnera un test de sudation pour déterminer si le bébé est atteint de la maladie. Si le test de chlorure de sueur est positif, le bébé est atteint de fibrose kystique et sera généralement dirigé vers un centre de soins de santé accrédité pour commencer le traitement. Si les tests génétiques révèlent une mutation dans le gène CFTR mais que le test de chlorure de sueur est négatif, le bébé est porteur de mucoviscidose mais n’a pas la maladie et ne nécessite pas de traitement. Dans les deux cas, un conseil génétique est généralement fait avec la famille pour expliquer les implications à long terme des résultats.

