Lorsque le cancer affecte les os, il est le plus souvent dû à un cancer non osseux qui a commencé ailleurs dans le corps et s’est propagé, ou métastasé, aux os. En revanche, l’accent est mis ici sur les cancers qui commencent dans les os, également connus sous le nom de cancers primaires des os.
Le cancer primaire des os est en réalité une vaste catégorie, constituée de nombreux types de tumeurs malignes, dont certaines sont très rares; cependant, parmi ceux-ci, l’ostéosarcome, le chondrosarcome et le sarcome d’Ewing sont parmi les plus communs.
Causes connues
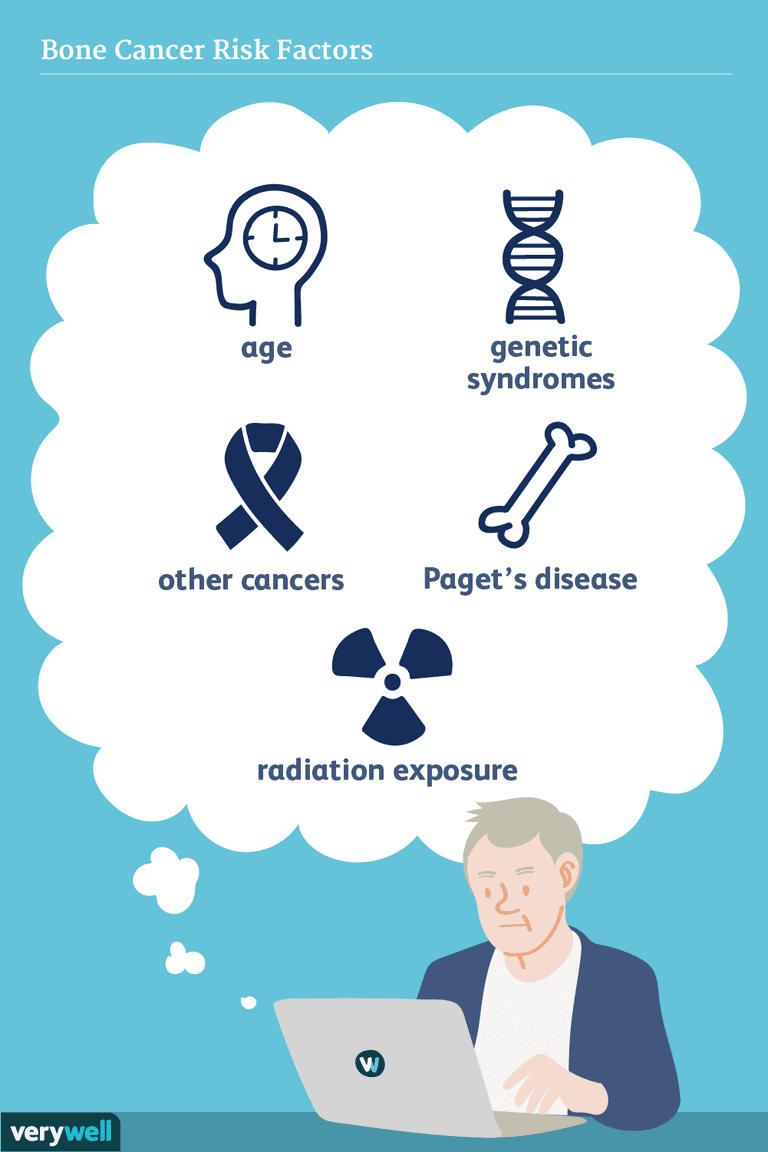
Bien que les causes du cancer des os ne soient pas connues avec précision, les changements dans l’ADN des cellules cancéreuses sont connus pour être importants. Dans la plupart des cas, ces changements se produisent par hasard et ne sont pas transmis des parents aux enfants.
Les scientifiques ont étudié les modèles de développement pour essayer de comprendre les facteurs de risque impliqués. L’ostéosarcome est le troisième type de malignité le plus fréquent qui affecte les os chez les adolescents, précédé seulement par la leucémie et le lymphome. Le chondrosarcome est également un cancer primitif osseux primaire, mais il est plus fréquent chez les adultes que chez les enfants et les adolescents, avec un âge moyen au diagnostic de 51 ans. Le sarcome d’Ewing est le plus souvent diagnostiqué chez les adolescents et l’âge moyen de diagnostic est de 15 ans.
Profil de risque de l’ostéosarcome
L’ostéosarcome est le cancer primaire des os le plus courant dans l’ensemble. Il y a quelques conditions spécifiques connues pour augmenter les chances de le développer. Les personnes qui ont une tumeur rare de l’œil connue sous le nom de rétinoblastome héréditaire ont un risque accru de développer un ostéosarcome.
En outre, ceux qui ont déjà été traités pour un cancer en utilisant la radiothérapie et la chimiothérapie ont un risque accru de développer un ostéosarcome plus tard dans la vie.
Incidemment, la plupart des médecins conviennent que les os cassés et blessés et les blessures sportives ne causent pas d’ostéosarcome. Cependant, de telles blessures peuvent amener un ostéosarcome déjà existant ou une autre tumeur osseuse à des soins médicaux, il y a donc certainement un lien entre les deux – c’est juste que la lésion mécanique ne semble pas causer l’ostéosarcome.
Facteurs de risque liés à l’âge, au sexe et à l’origine ethnique
L’ostéosarcome touche principalement deux groupes d’âge de pointe: le premier pic se situe dans l’adolescence et le deuxième chez les adultes plus âgés.
- Chez les patients plus âgés, l’ostéosarcome provient généralement d’un os anormal, comme ceux qui sont affectés par une maladie osseuse de longue date (par exemple, la maladie de Paget).
- Chez les personnes plus jeunes, l’ostéosarcome est extrêmement rare avant l’âge de cinq ans, et l’incidence du «pic» se produit en réalité pendant la poussée de croissance de l’adolescent. En moyenne, un «âge standard» représentatif de l’ostéosarcome chez les jeunes est de 16 ans pour les filles et de 18 ans pour les garçons. L’ostéosarcome est relativement rare par rapport à d’autres cancers; On estime que seulement environ 400 personnes de moins de 20 ans reçoivent un diagnostic d’ostéosarcome chaque année aux États-Unis. Les garçons sont plus souvent touchés dans la plupart des études, et l’incidence chez les jeunes d’ascendance africaine est légèrement plus élevée que chez les blancs. Facteurs de risque applicables aux plus jeunes
Présence de certains syndromes génétiques rares de cancer
Âge entre 10 et 30 ans
- Taille élevée
- Sexe masculin race Race afro-américaine
- Présence de certaines maladies osseuses
- Facteurs de risque applicables aux personnes âgées
- Certaines maladies osseuses comme la maladie de Paget, en particulier au fil du temps, sont associés à un risque accru d’ostéosarcome.
- Pourtant, le risque absolu est faible, avec seulement environ un pour cent des personnes atteintes de la maladie de Paget développant un ostéosarcome.
L’exposition aux rayonnements est un facteur de risque bien documenté et parce que l’intervalle entre l’irradiation pour le cancer et l’apparition de l’ostéosarcome est généralement plus long (par exemple, 10 ans ou plus). Pred Prédispositions génétiques
Les syndromes génétiques prédisposant à l’ostéosarcome comprennent:
Syndrome de Bloom an Anémie de Diamond-Blackfan
Syndrome de Li-Fraumeni
Maladie de Paget
Rétinoblastome
- Syndrome de Rothmund-Thomson (également appelé poikiloderma congénitale) syndrome Syndrome de Werner
- La fonction des gènes suppresseurs de tumeurs p53 et rétinoblastome joue un rôle important dans le développement de l’ostéosarcome.
- Bien que les mutations germinales (ovules et spermatozoïdes) des gènes p53 et rétinoblastome soient rares, ces gènes sont altérés dans la majorité des échantillons tumoraux d’ostéosarcome, il existe donc un lien avec le développement de l’ostéosarcome. Les mutations de la lignée germinale dans le gène p53 peuvent conduire à un risque élevé de développer des tumeurs malignes, y compris l’ostéosarcome qui a été décrit comme le syndrome de Li-Fraumeni.
- Bien que des altérations des gènes suppresseurs de tumeurs et des oncogènes soient nécessaires pour produire des ostéosarcomes, il n’est pas clair lequel de ces événements se produit en premier lieu et pourquoi ou comment il se produit.
- Les ostéosarcomes chez les personnes atteintes de la maladie de Paget
- Il existe un sous-ensemble rare d’ostéosarcomes dont le pronostic est très sombre. Les tumeurs ont tendance à survenir chez les personnes de plus de 60 ans. Les tumeurs sont grandes au moment de leur apparition et elles ont tendance à être très destructrices, rendant difficile une résection chirurgicale complète (retrait), et des métastases pulmonaires sont souvent présentes dès le début.
- Le profil de risque est celui d’un groupe d’âge plus avancé. Ils se développent chez environ un pour cent des personnes atteintes de la maladie de Paget, généralement lorsque de nombreux os sont touchés. Les tumeurs ont tendance à se produire dans la hanche, le fémur près des hanches, et dans l’os du bras près de l’articulation de l’épaule; ils sont difficiles à traiter chirurgicalement, principalement en raison de l’âge du patient et de la taille de la tumeur.
- L’amputation est parfois nécessaire, surtout lorsque l’os se brise à cause du cancer, ce qui est fréquent. Os Ostéosarcomes parostéens et périostés
Il s’agit d’un sous-ensemble qui est ainsi nommé en raison de sa localisation dans l’os; ce sont généralement des ostéosarcomes moins agressifs qui apparaissent à la surface de l’os en association avec la couche de tissu qui entoure l’os ou le périoste. Ils pénètrent rarement dans les parties internes de l’os et deviennent rarement des ostéosarcomes hautement malins.
Le profil de risque de l’ostéosarcome parostéal diffère de celui de l’ostéosarcome classique: il est plus fréquent chez les femmes que chez les hommes, il est plus fréquent chez les 20 à 40 ans et se manifeste généralement à l’arrière du fémur, près du genou articulaire, bien que tout os dans le squelette puisse être affecté. Pr Pronostic à risque plus élevé
Les facteurs de risque ont été associés à des pronostics de meilleure qualité mais, malheureusement, ces mêmes facteurs n’ont généralement pas permis d’identifier les patients susceptibles de bénéficier de régimes thérapeutiques plus intenses ou moins intenses. Les facteurs connus pour influencer les résultats sont les suivants. T Site tumoral primaire
Parmi les tumeurs qui se forment dans les bras et les jambes, celles qui sont plus éloignées du tronc ou du torse ont un meilleur pronostic. Tum Les tumeurs primaires qui se forment dans le crâne et la colonne vertébrale sont associées au plus grand risque de progression et de décès, principalement parce qu’il est plus difficile d’obtenir une élimination chirurgicale complète du cancer à ces endroits. Les ostéosarcomes de la tête et du cou dans la région de la mâchoire et de la bouche ont un meilleur pronostic que les autres sites primaires de la tête et du cou, peut-être parce qu’ils sont détectés plus tôt. Os Les ostéosarcomes d’Hipbone représentent sept à neuf pour cent de tous les ostéosarcomes; les taux de survie des patients sont de 20 à 47%.
Les patients atteints d’ostéosarcome multifocal (définis comme des lésions osseuses multiples sans tumeur primaire claire) ont un pronostic extrêmement mauvais.
Localisée par rapport à la maladie métastatique
Les patients atteints d’une maladie localisée (pas de propagation dans des régions éloignées) ont un bien meilleur pronostic que les patients atteints d’une maladie métastatique. Jusqu’à 20 pour cent des patients auront des métastases détectables sur les scans au moment du diagnostic, avec le poumon étant le site le plus commun. Le pronostic pour les patients atteints d’une maladie métastatique semble être déterminé en grande partie par les sites de métastases, le nombre de métastases et la résécabilité chirurgicale de la maladie métastatique.
Pour les personnes atteintes d’une maladie métastatique, le pronostic semble être meilleur avec moins de métastases pulmonaires et lorsque la maladie s’est propagée à un seul poumon, plutôt qu’aux deux poumons. N Nécrose tumorale après chimiothérapie n La nécrose tumorale fait ici référence à un tissu cancéreux «mort» à la suite d’un traitement.
Après une chimiothérapie et une intervention chirurgicale, le pathologiste évalue la nécrose tumorale dans la tumeur enlevée. Les patients présentant au moins 90% de nécrose dans la tumeur primaire après chimiothérapie ont un meilleur pronostic que les patients avec moins de nécrose.
Cependant, les chercheurs notent que moins de nécrose ne devrait pas être interprétée comme signifiant que la chimiothérapie a été inefficace; Les taux de guérison pour les patients avec peu ou pas de nécrose après chimiothérapie d’induction sont beaucoup plus élevés que les taux de guérison pour les patients qui ne reçoivent pas de chimiothérapie.
Chondrosarcome Profil de risque
Il s’agit d’une tumeur maligne des cellules productrices de cartilage, et il représente environ 20% de toutes les tumeurs osseuses primaires. Chondrosarcome peut survenir seul ou secondairement, dans ce qui est connu comme la "dégénérescence maligne" des tumeurs bénignes (telles que l’ostéochondrome ou l’enchondrome bénigne). Les facteurs de risque comprennent:
l’âge:
survient habituellement chez les personnes de plus de 40 ans; cependant, il se produit également dans le groupe d’âge plus jeune, et quand il le fait, il tend à être d’une malignité de qualité supérieure capable de métastases.
Sexe:
Se produit avec une fréquence presque égale chez les deux sexes.
Lieu:
Peut se produire dans n’importe quel os, mais il y a une tendance au développement dans l’os de la hanche et le fémur. Chondrosarcome peut survenir dans d’autres os plats, tels que l’omoplate, les côtes et le crâne.
Génétique:
Le syndrome des exostoses multiples (parfois appelé syndrome des ostéochondromes multiples) est une maladie héréditaire qui provoque de nombreuses bosses sur les os d’une personne, principalement constitués de cartilage. Les exostoses peuvent être douloureuses et entraîner des déformations osseuses et / ou des fractures. La maladie est génétique (causée par une mutation dans l’un des 3 gènes EXT1, EXT2 ou EXT3), et les patients atteints de cette maladie ont un risque accru de chondrosarcome.
Autres tumeurs bénignes: en Un enchondrome est une tumeur bénigne du cartilage qui se développe dans l’os. Les personnes qui obtiennent beaucoup de ces tumeurs ont une maladie appelée enchondromatose multiple. Ils ont également un risque accru de développer un chondrosarcome.
Profil de risque du sarcome d’Ewing
Cet
est beaucoup plus fréquent chez les blancs (non hispaniques ou hispaniques) et moins répandu chez les Américains d’origine asiatique et extrêmement rare chez les Afro-Américains. Les tumeurs cicatricielles peuvent survenir à tout âge, mais elles sont plus fréquentes chez les adolescents et moins fréquentes chez les jeunes adultes et les jeunes enfants. Ils sont rares chez les personnes âgées.
- Presque toutes les cellules tumorales Ewing ont des modifications qui impliquent le gène EWS, qui se trouve sur le chromosome 22. L’activation du gène EWS entraîne une prolifération des cellules et le développement de ce cancer, mais la manière exacte dont cela se produit n’est pas encore clair.

